
GHRP-2 10mg (3ml)
GHRP-2 is a research-grade synthetic growth hormone secretagogue studied for potent GH-releasing effects with minimal desensitization, making it valuable for HPA axis and somatotropic axis research. Researchers investigating growth hormone signaling and anabolic pathways rely on pharmaceutical-grade purity for reproducible results. Available at BLL Peptides — USA-made, rigorously tested. ✅ COA tested every batch✅ 98%+…
Description
GHRP-2: Complete Research Guide – Growth Hormone Releasing Peptide-2 Mechanisms, Secretagogue Research, and Endocrine Applications
Last updated: March 2026
Executive Summary
Growth Hormone Releasing Peptide-2 (GHRP-2), also known by its International Nonproprietary Name pralmorelin and its research designation KP-102, is a synthetic hexapeptide growth hormone secretagogue (GHS) that acts as a potent agonist of the growth hormone secretagogue receptor type 1a (GHS-R1a). Developed through the pioneering work of Cyril Bowers and Frank Momany in the 1980s and 1990s, GHRP-2 represents the culmination of iterative structure-activity optimization within the original GHRP series and is widely recognized as the most potent peptidyl GH secretagogue among its class.
The peptide's amino acid sequence is D-Ala-D-2-Nal-Ala-Trp-D-Phe-Lys-NH2, a hexapeptide incorporating three D-amino acid residues and one non-natural amino acid (D-2-naphthylalanine) that collectively confer enhanced receptor binding affinity, metabolic stability, and resistance to enzymatic degradation. Its molecular formula is C45H55N9O6, with a molecular weight of approximately 817.97 Daltons (CAS: 158861-67-7). GHRP-2 stimulates GH release from anterior pituitary somatotrophs through activation of the Gq/11-coupled GHS-R1a signaling cascade, producing robust, dose-dependent elevations in circulating growth hormone levels that synergize with endogenous growth hormone-releasing hormone (GHRH) signaling [1].
Unlike the highly selective ipamorelin, GHRP-2 produces measurable co-stimulation of cortisol and prolactin at GH-effective doses, reflecting its broader endocrine activation profile through the GHS-R1a receptor [2]. GHRP-2 also stimulates appetite through the ghrelin signaling pathway, consistent with its role as a ghrelin receptor agonist. Despite these additional endocrine effects, GHRP-2 remains a critically important research compound due to its superior GH-releasing potency, its well-characterized pharmacology spanning over three decades of investigation, and its unique regulatory status as the only GHRP approved for clinical diagnostic use — marketed in Japan as GHRP Kaken (pralmorelin) for the diagnosis of growth hormone deficiency [3].
GHRP-2's significance extends beyond its individual pharmacology. As the most potent member of the original GHRP family, it serves as the benchmark secretagogue against which newer compounds are evaluated, and its mechanisms of action have been instrumental in elucidating the fundamental biology of the GHS-R1a receptor and the ghrelin signaling axis.
Interactive 3D Molecular Structure
The following interactive 3D visualization renders the GHRP-2 hexapeptide in a ball-and-stick representation. Because GHRP-2 contains only six residues, each amino acid is displayed as a large sphere with connecting bonds, clearly illustrating the three D-amino acid modifications (D-Ala, D-2-Nal, D-Phe) that are critical for receptor binding and metabolic stability, as well as the aromatic tryptophan (Trp) residue essential for GHS-R1a activation.
Legend: The interactive visualization above depicts GHRP-2's six-residue structure in a ball-and-stick representation. The three D-amino acid / non-natural residues — D-Ala (D-alanine), D-2-Nal (D-2-naphthylalanine), and D-Phe (D-phenylalanine) — are highlighted in purple, reflecting their critical role in conferring receptor affinity and proteolytic resistance. Tryptophan is shown in cyan (aromatic), alanine in gray-blue (hydrophobic), lysine in red (positively charged), and the C-terminal amide (NH2) in gold. Drag to rotate the structure; scroll to zoom.
Table of Contents
- Introduction and Development History
- Molecular Structure and Chemistry
- Detailed Mechanism of Action
- Scientific Research Review
- Clinical Investigations and Diagnostic Use
- Comparison with Other GH Secretagogues
- Pharmacokinetics and Safety Profile
- Research Applications
- References
- Disclaimer
Introduction and Development History
The Birth of Growth Hormone Secretagogues
The discovery of growth hormone-releasing peptides traces back to the seminal work of Cyril Y. Bowers at Tulane University in the late 1970s and early 1980s. Bowers and his colleague Frank Momany at the University of Texas demonstrated that synthetic analogs of Met-enkephalin could stimulate growth hormone release from anterior pituitary cells through a mechanism entirely independent of the growth hormone-releasing hormone (GHRH) receptor [1]. This landmark observation revealed the existence of a previously unknown GH regulatory pathway and initiated a new era in neuroendocrine pharmacology.
The first compound to emerge from this program was growth hormone-releasing peptide-6 (GHRP-6), a hexapeptide with the sequence His-D-Trp-Ala-Trp-D-Phe-Lys-NH2. While GHRP-6 demonstrated potent GH-releasing activity both in vitro and in vivo, it also exhibited significant limitations, including stimulation of appetite, elevation of cortisol and prolactin, and only moderate receptor binding affinity [4]. These shortcomings provided the impetus for systematic structure-activity relationship (SAR) studies aimed at optimizing the GHRP pharmacophore.
Development of GHRP-2: Optimization of the GHRP Scaffold
GHRP-2 was developed through iterative chemical modification of the GHRP-6 scaffold by Bowers and Momany during the late 1980s. The key structural innovation was the replacement of the N-terminal His-D-Trp dipeptide in GHRP-6 with D-Ala-D-2-Nal, where D-2-Nal represents D-2-naphthylalanine, a non-natural aromatic amino acid featuring an extended bicyclic naphthalene ring system [5]. This modification produced dramatic improvements in both receptor binding affinity and GH-releasing potency.
The rationale for incorporating D-2-naphthylalanine centered on enhancing hydrophobic interactions within the GHS-R1a binding pocket. The naphthalene ring system provides approximately 40% greater hydrophobic surface area compared to the indole ring of tryptophan, enabling stronger van der Waals contacts with hydrophobic residues lining the receptor's transmembrane binding cavity [6]. Combined with the D-configuration that confers resistance to aminopeptidases and chymotrypsin-like proteases, this substitution yielded a compound with both superior potency and improved metabolic stability.
Comparative studies established that GHRP-2 was the most potent GH secretagogue among the original GHRP series, with approximately 2-3 fold greater GH-releasing activity than GHRP-6 and hexarelin on a molar basis in human studies [7]. This potency advantage, combined with a well-characterized safety profile from extensive preclinical and clinical investigation, established GHRP-2 as the reference standard for the GHRP class.
Regulatory Milestones: Approval in Japan
GHRP-2 achieved a unique regulatory distinction when it was approved in Japan in 2004 under the trade name GHRP Kaken (pralmorelin dihydrochloride) for use as a diagnostic agent in the evaluation of growth hormone deficiency (GHD). Manufactured by Kaken Pharmaceutical Co., Ltd., the product is administered as an intravenous bolus injection (100 micrograms), and the subsequent GH response is measured at defined time points to assess pituitary somatotroph reserve [3]. This diagnostic application exploits the predictable, robust GH response produced by GHRP-2 in subjects with intact pituitary function, with a blunted or absent response indicating GHD.
The approval of pralmorelin in Japan represents the only regulatory authorization for a GHRP as a diagnostic or therapeutic agent in any major market. Clinical validation studies in Japanese populations demonstrated that GHRP-2 produced superior diagnostic sensitivity and specificity for GHD compared to the traditional insulin tolerance test (ITT), with the added advantage of being simpler to administer and carrying no risk of symptomatic hypoglycemia [8]. International studies subsequently confirmed these findings, with Mahajan and colleagues reporting that the GHRP-2 stimulation test achieved 100% sensitivity and 100% specificity for severe GHD when using an appropriate GH cutoff value [9].
Nomenclature and Classification
GHRP-2 is classified within the growth hormone secretagogue (GHS) family as a peptidyl ghrelin receptor (GHS-R1a) agonist. Its established nomenclature includes:
- INN (International Nonproprietary Name): Pralmorelin
- Research designations: KP-102, GHRP-2
- Trade name: GHRP Kaken (Japan)
- Chemical classification: Synthetic hexapeptide growth hormone secretagogue
The compound should be distinguished from other GHRP family members including GHRP-6 (His-D-Trp-Ala-Trp-D-Phe-Lys-NH2), hexarelin (His-D-2-Me-Trp-Ala-Trp-D-Phe-Lys-NH2), and ipamorelin (Aib-His-D-2-Nal-D-Phe-Lys-NH2), as well as the non-peptidyl GHS MK-677 (ibutamoren).
Molecular Structure and Chemistry
Amino Acid Sequence
GHRP-2 is a linear hexapeptide with the following primary sequence:
D-Ala-D-2-Nal-Ala-Trp-D-Phe-Lys-NH2
This six-residue peptide incorporates four strategically positioned modifications relative to a hypothetical all-L natural amino acid peptide:
-
D-Ala (D-alanine, position 1): The D-enantiomer of alanine at the N-terminus provides resistance to aminopeptidases, which preferentially cleave L-amino acids from the peptide N-terminus. D-Ala's small side chain minimizes steric interference with downstream receptor contacts while ensuring metabolic stability. This position was occupied by L-His in the parent compound GHRP-6; the switch to D-Ala removed unwanted histamine-related effects and improved the potency profile [5].
-
D-2-Nal (D-2-naphthylalanine, position 2): The signature residue of GHRP-2, D-2-naphthylalanine features an extended bicyclic aromatic ring system (naphthalene) in place of the single-ring indole moiety of tryptophan found at the analogous position in GHRP-6. The 2-naphthyl substituent provides an enlarged pi-electron surface that engages in enhanced aromatic stacking and hydrophobic interactions with the GHS-R1a transmembrane domain. The D-configuration simultaneously confers enzymatic resistance and optimizes the spatial orientation for receptor binding [6, 10].
-
D-Phe (D-phenylalanine, position 5): Conserved across all GHRP family members, D-Phe at position 5 is critical for GHS-R1a binding. SAR studies demonstrated that substitution with L-Phe or removal of this residue abolishes GH-releasing activity, indicating that the D-configuration is essential for correct pharmacophoric geometry [5].
-
C-terminal amidation (Lys-NH2): The amidation of the C-terminal lysine residue neutralizes the carboxylate negative charge, enhances hydrogen bonding capacity with receptor residues, and improves membrane permeability. Removal of the amide group reduces GH-releasing potency by approximately 10-fold [1].
The remaining two residues — L-Ala at position 3 and L-Trp at position 4 — retain their natural L-configuration. Tryptophan at position 4 is highly conserved across GHRPs and contributes an indole ring that is essential for receptor activation, likely through aromatic interactions with Phe279 and Phe312 in the GHS-R1a transmembrane domain [11].
Physicochemical Properties
| Property | Value |
|---|---|
| Molecular Formula | C45H55N9O6 |
| Molecular Weight | Approximately 817.97 Da |
| CAS Number | 158861-67-7 |
| Sequence | D-Ala-D-2-Nal-Ala-Trp-D-Phe-Lys-NH2 |
| Sequence Length | 6 amino acids (hexapeptide) |
| Appearance | White to off-white lyophilized powder |
| Solubility | Freely soluble in water, DMSO; soluble in dilute acetic acid |
| pI (Isoelectric Point) | Approximately 9.4 |
| Net Charge at pH 7.4 | +1 (from Lys epsilon-amino group) |
| Storage | -20C lyophilized; 2-8C reconstituted (use within 21 days) |
| Purity (Research Grade) | Greater than 98% by HPLC |
Structural Comparison with GHRP-6 and Ghrelin
GHRP-2 bears significant structural homology with its parent compound GHRP-6, sharing four of six residues (Ala3, Trp4, D-Phe5, Lys6) with identical stereochemistry. The two N-terminal substitutions (D-Ala for His, D-2-Nal for D-Trp) are entirely responsible for GHRP-2's enhanced potency and modified endocrine profile. In contrast, GHRP-2 bears no primary sequence homology with ghrelin, the endogenous 28-amino acid ligand of GHS-R1a. Ghrelin requires an octanoyl modification on Ser3 for receptor activation, whereas GHRP-2 achieves receptor engagement through an entirely different set of hydrophobic and aromatic pharmacophoric elements [12].
Despite these structural differences, both GHRP-2 and ghrelin converge on the same orthosteric binding site of GHS-R1a, with computational modeling studies suggesting that the D-2-Nal and Trp residues of GHRP-2 occupy spatial positions analogous to the octanoyl chain and Phe4 of ghrelin, respectively [11]. This convergent pharmacophore occupancy explains the competitive binding relationship observed between GHRPs and ghrelin in radioligand displacement assays.
Detailed Mechanism of Action
GHS-R1a Receptor Activation
GHRP-2's primary mechanism of action involves potent agonism of the growth hormone secretagogue receptor type 1a (GHS-R1a), a seven-transmembrane G protein-coupled receptor (GPCR) that was identified by Howard and colleagues in 1996 through expression cloning using MK-0677 as the probe ligand [13]. The GHS-R1a receptor is expressed predominantly on somatotroph cells of the anterior pituitary gland, with additional expression in hypothalamic arcuate nucleus neurons, vagal afferent neurons, and various peripheral tissues including the pancreas, adrenal glands, and gastrointestinal tract.
The signaling cascade initiated by GHRP-2 at pituitary somatotrophs proceeds through the following steps:
-
Receptor binding: GHRP-2 engages the orthosteric binding pocket of GHS-R1a within the transmembrane helical bundle. Key molecular contacts include hydrophobic interactions between the D-2-Nal naphthalene ring and residues Phe279 (TM6), Phe312 (TM7), and Ile178 (ECL2), as well as a salt bridge between the protonated Lys6 epsilon-amino group and Glu124 (TM3) [11].
-
Gq/11 coupling and PLC activation: Ligand-induced conformational change in GHS-R1a promotes coupling to Gq/11 heterotrimeric G proteins. Activated Gq/11 stimulates phospholipase C-beta (PLC-beta), which hydrolyzes phosphatidylinositol 4,5-bisphosphate (PIP2) into inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG) [14].
-
Intracellular calcium mobilization: IP3 binds to IP3 receptors on the endoplasmic reticulum membrane, triggering release of stored calcium ions into the cytoplasm. DAG concurrently activates protein kinase C (PKC), which phosphorylates and activates voltage-gated L-type calcium channels at the plasma membrane, producing additional calcium influx from the extracellular space.
-
GH vesicle exocytosis: The combined calcium release from intracellular stores and extracellular influx produces a robust elevation in cytoplasmic calcium concentration that triggers the SNARE-mediated fusion of GH-containing secretory granules with the somatotroph plasma membrane, resulting in pulsatile GH release into the pituitary portal circulation [14].
Hypothalamic Actions
In addition to its direct pituitary effects, GHRP-2 exerts significant actions at the hypothalamic level. GHS-R1a is expressed on neuropeptide Y (NPY) and agouti-related peptide (AgRP) neurons in the arcuate nucleus, as well as on GHRH-producing neurons of the arcuate and ventromedial nuclei. GHRP-2 activates these hypothalamic populations to produce several complementary effects:
-
GHRH release stimulation: GHRP-2 activates GHS-R1a on GHRH neurons, stimulating the release of endogenous GHRH into the hypophyseal portal circulation. This GHRH then acts on pituitary GHRH receptors (a Gs/cAMP-coupled pathway), synergizing with GHRP-2's direct Gq/calcium signaling to produce amplified GH release [15].
-
Somatostatin suppression: Evidence from hypothalamic explant studies indicates that GHRPs can functionally antagonize somatostatin release from periventricular neurons, effectively releasing the somatotroph from tonic inhibitory control. This disinhibition further amplifies the GH response [16].
-
Appetite stimulation: Activation of GHS-R1a on arcuate NPY/AgRP neurons stimulates the orexigenic neuropeptide pathway, producing appetite stimulation and increased food intake. This effect is mediated through the same receptor system engaged by endogenous ghrelin and is a consistent pharmacological feature of GHRP-2 that distinguishes it from the more selective ipamorelin [17].
Synergy with GHRH Pathway
A critical pharmacological property of GHRP-2 is its synergistic interaction with GHRH signaling. When GHRP-2 and GHRH (or GHRH analogs such as CJC-1295 or sermorelin) are co-administered, the resulting GH response is substantially greater than the sum of individual responses. This synergy arises from the convergence of two distinct intracellular signaling cascades on the somatotroph:
-
GHRH pathway: GHRH-R activation leads to Gs-mediated stimulation of adenylyl cyclase, elevation of cAMP, activation of protein kinase A (PKA), and PKA-dependent phosphorylation of L-type calcium channels and CREB transcription factor (which upregulates GH gene expression).
-
GHRP-2 pathway: GHS-R1a activation leads to Gq/11-mediated PLC activation, IP3-dependent calcium release, and PKC-dependent potentiation of calcium channels.
The simultaneous activation of cAMP/PKA and calcium/PKC signaling pathways produces a multiplicative effect on calcium-dependent exocytosis. Arvat and colleagues demonstrated in human studies that combined GHRP-2 plus GHRH administration produced GH peaks that were 2-3 fold greater than either agent alone, establishing this combination as the most potent known stimulus for GH release [7, 18].
Cortisol and Prolactin Co-Release
Unlike the highly selective ipamorelin, GHRP-2 produces measurable elevations in both cortisol and prolactin at GH-effective doses. The cortisol response is mediated through stimulation of ACTH release from anterior pituitary corticotrophs, likely through GHS-R1a expressed on these cells or through hypothalamic CRH neuron activation [2]. The prolactin response is thought to involve activation of lactotroph GHS-R1a or indirect modulation through hypothalamic dopamine pathways.
Importantly, the cortisol and prolactin responses to GHRP-2 are dose-dependent but proportionally smaller than the GH response, and they remain within physiological ranges at standard research doses. Arvat and colleagues reported that GHRP-2 at 1 microgram/kg IV produced significant GH elevation with modest cortisol increases, while higher doses (2-3 micrograms/kg) produced proportionally greater cortisol and prolactin responses [7]. This dose-dependent broadening of the endocrine response distinguishes GHRP-2 from ipamorelin, which maintains GH selectivity across its full dose range, and from GHRP-6, which produces relatively larger cortisol responses at equivalent GH-effective doses.
Somatostatin Sensitivity
Like other peptidyl GH secretagogues, GHRP-2-stimulated GH release remains sensitive to somatostatin (SRIF) inhibition. When somatostatin tone is elevated — as occurs physiologically following a GH pulse or pharmacologically through exogenous somatostatin infusion — GHRP-2's efficacy is attenuated. This somatostatin sensitivity preserves the pulsatile pattern of GH secretion that is essential for normal physiological responses, including hepatic IGF-1 production, linear bone growth, and metabolic regulation [16]. This property contrasts with the non-peptidyl GHS MK-677, which partially overrides somatostatin inhibition, potentially leading to more continuous (rather than pulsatile) GH elevation.
Scientific Research Review
In Vitro Studies: Receptor Binding and Signaling
Foundational in vitro studies established the pharmacological profile of GHRP-2 at the molecular level. Using cloned human GHS-R1a expressed in HEK293 cells, Bednarek and colleagues determined that GHRP-2 binds to GHS-R1a with a Ki of approximately 1.2 nM, representing one of the highest binding affinities among peptidyl GHS compounds. In comparison, GHRP-6 exhibited a Ki of approximately 3.4 nM and hexarelin approximately 2.8 nM in the same assay system [6, 10].
Functional assays measuring intracellular calcium mobilization in GHS-R1a-expressing cells demonstrated that GHRP-2 acts as a full agonist with an EC50 of approximately 0.9 nM, achieving maximal efficacy (Emax) comparable to ghrelin. These binding and functional data confirmed GHRP-2 as the most potent peptidyl agonist in the GHRP series [10].
Studies in primary rat pituitary cell cultures demonstrated that GHRP-2 stimulated GH release in a concentration-dependent manner (EC50 approximately 10 nM) and that this effect was completely abolished by the GHS-R1a antagonist D-Lys3-GHRP-6, confirming receptor dependence. Co-incubation with GHRH produced synergistic GH release, with the combination exceeding the sum of individual responses by approximately 2.5-fold [19].
In Vivo Animal Studies
Rodent GH Release Studies: Extensive dose-response characterization in rats established that GHRP-2 administered subcutaneously at doses of 25-200 micrograms/kg produced dose-dependent GH peaks, with maximal responses occurring at approximately 15-20 minutes post-injection and return to baseline within 60-90 minutes. The ED50 for GH stimulation in rats was approximately 50 micrograms/kg SC [5].
Chronic Administration and Growth Effects: Bowers and colleagues demonstrated that repeated daily GHRP-2 administration (100 micrograms/kg SC, twice daily) for 14 days in young rats produced significant increases in body weight gain (approximately 18% above controls), circulating IGF-1 levels (approximately 35% elevation), and tibial epiphyseal plate width, confirming sustained somatotropic axis activation without tachyphylaxis [1, 5].
Adiposity and Body Composition: Research by Tschop and colleagues revealed that chronic GHRP-2 administration in rodents increased food intake and produced a biphasic effect on body composition: initial increases in both lean and fat mass followed by a predominant lean mass accrual at later time points, consistent with the dual orexigenic and GH-mediated lipolytic effects of GHS-R1a agonism [17].
Cardiovascular Research: Nagaya and colleagues investigated the effects of GHRP-2 in a rat model of heart failure and demonstrated that chronic GHRP-2 administration improved left ventricular function, reduced cardiac fibrosis, and attenuated the progression of cardiac cachexia. These cardioprotective effects were attributed to both direct GHS-R1a-mediated actions on cardiomyocytes and indirect effects through GH/IGF-1 axis activation [20].
Neuroprotective Studies: Research by Frago and colleagues demonstrated that GHRP-2 exerted neuroprotective effects in rodent models of brain injury, reducing apoptotic cell death in hippocampal neurons and preserving cognitive function following ischemic insult. These effects appeared to involve both GHS-R1a-dependent and GHS-R1a-independent mechanisms, with evidence for direct anti-apoptotic signaling through PI3K/Akt pathway activation [21].
Human Pharmacology Studies
Dose-Response Characterization: Arvat and colleagues at the University of Turin conducted pivotal human pharmacology studies characterizing the dose-response relationship of GHRP-2 for GH, ACTH, cortisol, and prolactin release. In healthy adult volunteers, intravenous GHRP-2 at doses of 0.1, 0.3, 1.0, and 3.0 micrograms/kg produced dose-dependent GH increases with peak concentrations at 15-30 minutes. The 1.0 microgram/kg dose produced a mean GH peak of approximately 50-80 ng/mL, while cortisol showed modest elevations of approximately 20-40% above baseline. Prolactin responses were detectable but smaller in magnitude than the GH response [7].
Aging and GH Response: Bowers and colleagues demonstrated that GHRP-2 partially overcomes the age-related decline in GH secretory capacity (somatopause). Elderly subjects (65-80 years) receiving GHRP-2 at 1 microgram/kg IV showed GH responses that were reduced compared to young adults but still substantially above baseline, indicating preserved GHS-R1a receptor function and responsiveness despite age-related reductions in somatotroph number and GHRH receptor density [22].
GHRP-2 Plus GHRH Synergy in Humans: The synergistic interaction between GHRP-2 and GHRH has been extensively characterized in human studies. Arvat and colleagues reported that combined administration of GHRP-2 (1 microgram/kg IV) and GHRH (1 microgram/kg IV) produced mean GH peaks of approximately 90-150 ng/mL, representing a 2-3 fold amplification over either agent alone. This combination was described as the most potent known stimulus for GH release in humans and has been proposed as a superior diagnostic test for GH reserve assessment [7, 18].
Metabolic Effects: Cordido and colleagues investigated the metabolic effects of GHRP-2 in obese human subjects and demonstrated that GHRP-2 administration produced significant GH release even in the setting of obesity-associated GH suppression. Furthermore, GHRP-2 enhanced the GH response to GHRH in obese individuals, partially overcoming the GH refractoriness associated with elevated body mass index, suggesting potential research applications in metabolic physiology [23].
Clinical Investigations and Diagnostic Use
GH Deficiency Diagnosis: The Pralmorelin Stimulation Test
The primary clinical application of GHRP-2 is as a diagnostic provocative agent for the assessment of pituitary GH secretory capacity. The GHRP-2 stimulation test (also called the pralmorelin test) is performed as follows:
- The subject fasts overnight (minimum 8 hours)
- Baseline blood samples are drawn for GH measurement
- GHRP-2 (pralmorelin) 100 micrograms is administered as an intravenous bolus
- Serial blood samples are collected at 15, 30, 45, 60, and 90 minutes post-injection
- Peak GH concentration is compared against established diagnostic cutoff values
In the Japanese regulatory submission, a peak GH response below 9 ng/mL following GHRP-2 stimulation was used as the diagnostic criterion for severe GHD, with sensitivity and specificity exceeding 90% [3]. International validation studies have generally confirmed this performance. Mahajan and Lightman reported in 2002 that the GHRP-2 test achieved 100% sensitivity and 100% specificity for severe GHD when using a peak GH cutoff of 15 micrograms/L in a European adult population, outperforming the insulin tolerance test (ITT) in practical utility [9].
The GHRP-2 stimulation test offers several practical advantages over the traditional ITT:
- Safety: No risk of symptomatic hypoglycemia, seizures, or cardiovascular events associated with insulin-induced hypoglycemia
- Simplicity: Single bolus injection versus careful insulin dose titration
- Reproducibility: More consistent GH responses with lower intra-subject variability
- Patient comfort: Well-tolerated with minimal side effects (transient flushing, mild hunger)
- Contraindication profile: Fewer contraindications than ITT (which is contraindicated in seizure disorders, ischemic heart disease, and elderly patients) [8, 9]
Combined GHRP-2 + GHRH Diagnostic Test
To further enhance diagnostic sensitivity, researchers have proposed a combined GHRP-2 plus GHRH stimulation test. Popovic and colleagues evaluated this combination and demonstrated that co-administration of GHRP-2 (1 microgram/kg IV) and GHRH (1 microgram/kg IV) produced the maximal possible GH response, providing a definitive assessment of maximal pituitary somatotroph capacity. A severely blunted response to this combination was considered diagnostic of severe organic GHD, as it eliminates the possibility of a false-negative result due to inadequate hypothalamic stimulation [24].
Investigational Therapeutic Applications
Beyond diagnostic use, GHRP-2 has been investigated in several therapeutic research contexts:
Growth Hormone Deficiency: Pihoker and colleagues evaluated GHRP-2 as a potential therapeutic agent for GH-deficient children and demonstrated sustained GH secretory responses over treatment periods of several weeks, with consequent increases in IGF-1 and growth velocity. However, the requirement for frequent injections (due to GHRP-2's short duration of action) limited practical therapeutic development compared to recombinant GH [25].
Cachexia and Wasting: The combination of GH-releasing and appetite-stimulating properties makes GHRP-2 a theoretically attractive agent for cachectic conditions. Preclinical studies in models of cancer cachexia, chronic kidney disease-associated wasting, and HIV-associated wasting have demonstrated that GHRP-2 administration improved food intake, lean body mass, and functional outcomes [20].
Cardiac Function: Building on the preclinical work of Nagaya and colleagues, clinical interest has emerged in the cardioprotective potential of GHS-R1a agonism. GHRP-2 and related GH secretagogues have been shown to improve cardiac contractility, reduce ischemia-reperfusion injury, and attenuate pathological remodeling in experimental heart failure models [20].
Comparison with Other GH Secretagogues
GHRP Family Comparison Table
| Property | GHRP-2 | GHRP-6 | Hexarelin | Ipamorelin |
|---|---|---|---|---|
| Sequence | D-Ala-D-2Nal-Ala-Trp-D-Phe-Lys-NH2 | His-D-Trp-Ala-Trp-D-Phe-Lys-NH2 | His-D-2MeTrp-Ala-Trp-D-Phe-Lys-NH2 | Aib-His-D-2Nal-D-Phe-Lys-NH2 |
| Residues | 6 | 6 | 6 | 5 |
| MW (Da) | 817.97 | 873.01 | 887.04 | 711.85 |
| GH Potency (relative) | Highest | Moderate | High | Moderate |
| GH Selectivity | Moderate | Low | Low | Highest |
| Cortisol Stimulation | Moderate | Significant | Moderate | None at GH doses |
| Prolactin Stimulation | Moderate | Moderate | Significant | None at GH doses |
| Appetite Stimulation | Moderate | Significant | Mild | Minimal |
| Receptor | GHS-R1a | GHS-R1a | GHS-R1a | GHS-R1a |
| Clinical Status | Approved (Japan, diagnostic) | Research only | Research only | Phase II (discontinued) |
GHS-R1a Agonist Comparison: Peptidyl vs. Non-Peptidyl
| Property | GHRP-2 | Ipamorelin | MK-677 (Ibutamoren) |
|---|---|---|---|
| Chemical Class | Hexapeptide | Pentapeptide | Non-peptidyl (spiroindoline) |
| Route | SC / IV injection | SC / IV injection | Oral |
| Half-life | Approximately 25-30 min | Approximately 2 hours | Approximately 4-6 hours |
| Duration of GH elevation | 1-2 hours | 1-3 hours | 8-12 hours |
| GH pattern | Pulsatile | Pulsatile | Sustained/elevated trough |
| Somatostatin sensitivity | Preserved | Preserved | Partially overridden |
| Cortisol elevation | Yes (moderate) | No | Yes (mild-moderate) |
| Prolactin elevation | Yes (moderate) | No | Yes (mild) |
| IGF-1 elevation | Yes (dose-dependent) | Yes (dose-dependent) | Yes (sustained) |
| Appetite stimulation | Moderate | Minimal | Significant |
| Regulatory status | Approved (Japan) | Research | Research |
GHRP-2 vs. GHRH Analogs
| Property | GHRP-2 | CJC-1295 | Sermorelin |
|---|---|---|---|
| Receptor Target | GHS-R1a (ghrelin receptor) | GHRH-R | GHRH-R |
| Signaling Pathway | Gq/11 — PLC — IP3/Ca2+ | Gs — AC — cAMP/PKA | Gs — AC — cAMP/PKA |
| GH Release Mechanism | Calcium-dependent exocytosis + GHRH potentiation | cAMP-dependent GH synthesis and release | cAMP-dependent GH synthesis and release |
| Synergy | Synergizes with GHRH analogs | Synergizes with GHRPs | Synergizes with GHRPs |
| Appetite Effect | Stimulatory (ghrelin-mimetic) | None | None |
| Cortisol Effect | Moderate elevation | None | None |
| Half-life | Approximately 25-30 min | Approximately 30 min (without DAC) | Approximately 12 min |
| Clinical Use | GHD diagnosis (Japan) | Research | Previously FDA-approved (diagnostic) |
Key Interpretive Notes
The comparison data above illustrate a fundamental trade-off within the GHS pharmacological space: GHRP-2 delivers the highest raw GH-releasing potency of any peptidyl secretagogue but at the cost of moderate cortisol and prolactin co-stimulation. In contrast, ipamorelin achieves the cleanest selectivity profile (GH-specific with no cortisol or prolactin effects) but with somewhat lower maximal GH release. Researchers must select the appropriate secretagogue based on their specific experimental requirements, balancing potency against selectivity.
For research protocols where maximal GH stimulation is the primary objective and moderate cortisol/prolactin co-stimulation is acceptable or even desirable (as in studies of the complete GHS endocrine response), GHRP-2 remains the peptide of choice. For protocols requiring isolated GH stimulation without confounding cortisol or prolactin variables, ipamorelin is the preferred agent [2, 7].
Pharmacokinetics and Safety Profile
Pharmacokinetic Parameters
GHRP-2 is administered via subcutaneous or intravenous injection. Following IV bolus administration in human subjects, the pharmacokinetic profile is characterized by:
- Tmax (GH peak): 15-30 minutes post-injection
- Duration of GH elevation: 60-120 minutes (return to baseline)
- Estimated plasma half-life: Approximately 25-30 minutes
- Bioavailability (SC): Approximately 60-70% relative to IV
- Clearance: Primarily through renal and hepatic peptidase-mediated degradation
- Volume of distribution: Consistent with extracellular fluid space
The relatively short plasma half-life of GHRP-2 necessitates repeated dosing to maintain sustained GH axis activation. This pharmacokinetic limitation was a key factor driving the development of longer-acting GHS compounds, including DAC-conjugated GHRH analogs (e.g., CJC-1295 DAC) and oral non-peptidyl agents (MK-677) [14].
Safety Profile
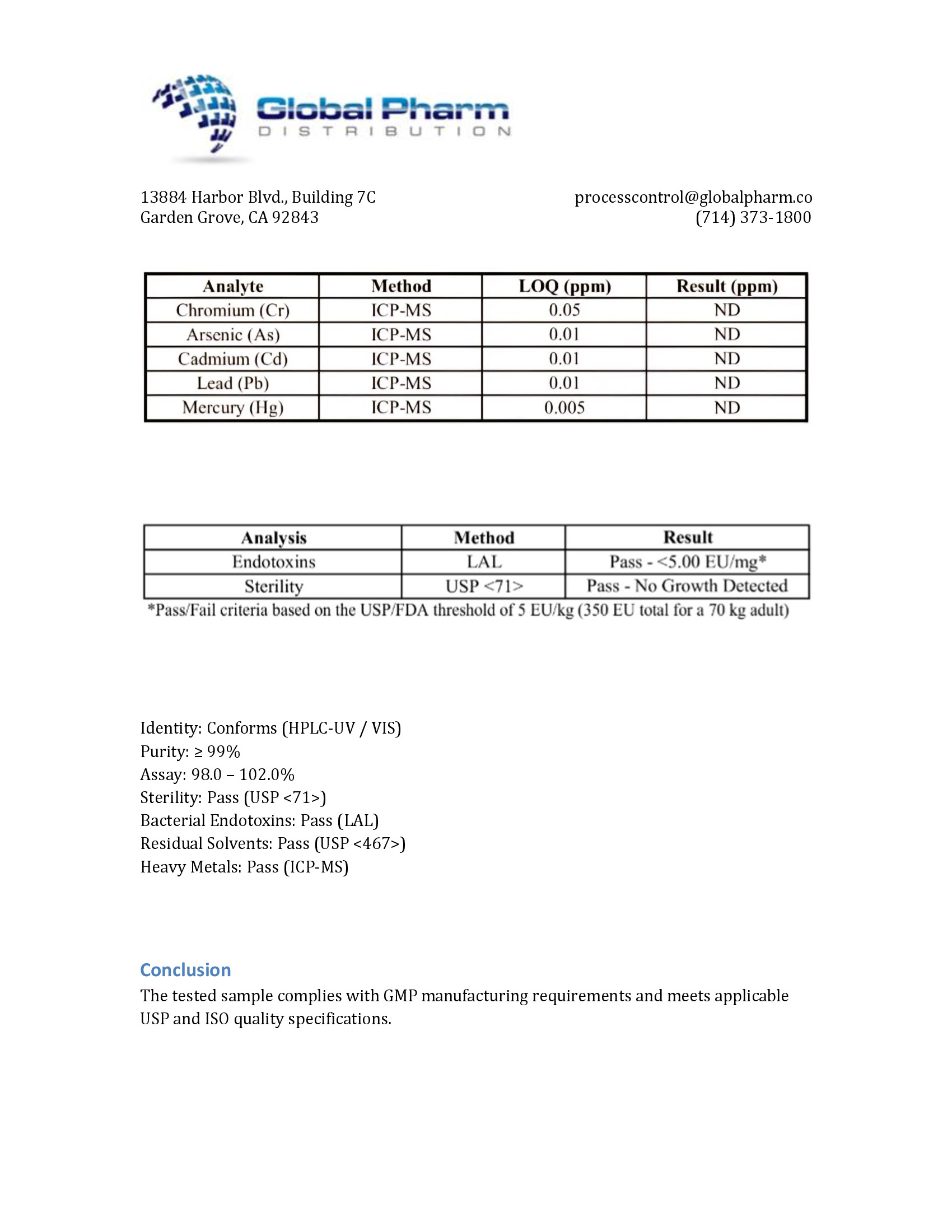
The safety of GHRP-2 has been extensively evaluated in both preclinical and clinical studies:
Acute Tolerability: In human pharmacology studies, single IV doses of GHRP-2 up to 3 micrograms/kg were well-tolerated, with the most commonly reported effects including transient facial flushing (approximately 20-30% of subjects), mild hunger sensation (consistent with ghrelin-mimetic activity), and occasional transient dizziness. No serious adverse events were reported at standard research doses [7, 8].
Endocrine Safety: While GHRP-2 elevates cortisol and prolactin, these elevations are dose-dependent, transient (returning to baseline within 1-2 hours), and remain within physiological ranges at standard doses. No evidence of sustained HPA axis activation, hypercortisolism, or hyperprolactinemia has been reported with repeated GHRP-2 administration in clinical studies [2, 7].
Metabolic Safety: No clinically significant effects on fasting glucose, insulin sensitivity, or lipid profiles have been reported in short-term human studies. The GH responses produced by GHRP-2 are somatostatin-sensitive and pulsatile, reducing the risk of the sustained GH elevation-associated insulin resistance that can occur with continuous GH or long-acting GHS exposure [8].
Preclinical Toxicology: Standard preclinical safety assessments in rodents and dogs revealed:
- No target organ toxicity at doses up to 100-fold the pharmacologically active dose
- No mutagenic potential in standard genotoxicity assays (Ames test, chromosomal aberration assay)
- No evidence of carcinogenic potential in chronic exposure studies
- Adequate safety margins between efficacious and toxic dose ranges [3]
Tachyphylaxis: Repeated administration of GHRP-2 can produce partial desensitization of the GH response over time, a phenomenon common to all GHS-R1a agonists. Studies in humans demonstrated that continuous infusion of GHRP-2 produced progressive attenuation of GH pulses, while intermittent bolus dosing (e.g., 2-3 times daily with intervals of 3-4 hours) maintained robust GH responses without significant tachyphylaxis [16, 22]. This observation supports the use of pulsatile rather than continuous dosing strategies in research protocols.
Adverse Effect Comparison Across GHRPs
| Adverse Effect | GHRP-2 | GHRP-6 | Hexarelin | Ipamorelin |
|---|---|---|---|---|
| Facial flushing | Occasional | Common | Occasional | Rare |
| Appetite increase | Moderate | Significant | Mild | Minimal |
| Cortisol elevation | Moderate | Significant | Moderate | None |
| Prolactin elevation | Moderate | Moderate | Significant | None |
| Water retention | Mild | Mild-moderate | Mild | Minimal |
| Paresthesias | Rare | Rare | Rare | Rare |
| Injection site reactions | Mild, transient | Mild, transient | Mild, transient | Mild, transient |
Research Applications
GHRP-2 serves as a versatile and critically important research tool across multiple domains of biomedical investigation:
1. Neuroendocrine Pharmacology and GH Axis Research
As the most potent peptidyl GH secretagogue, GHRP-2 is the preferred agent for studies requiring maximal GH stimulation. Its well-characterized dose-response curve and predictable endocrine profile make it an ideal pharmacological probe for investigating somatotroph function, GH pulse dynamics, and the interaction between GHS-R1a and GHRH signaling pathways. The simultaneous cortisol and prolactin co-stimulation, while a limitation for isolated GH studies, provides additional information about corticotroph and lactotroph function in integrated neuroendocrine assessments [7, 18].
2. Diagnostic Assessment of GH Deficiency
GHRP-2's regulatory approval in Japan as a GHD diagnostic agent validates its utility for clinical research into growth hormone disorders. The pralmorelin stimulation test has been validated against the insulin tolerance test, arginine stimulation test, and combined GHRH-arginine test as a reliable, safe, and reproducible method for assessing pituitary GH reserve [3, 8, 9].
3. Aging and Somatopause Research
The age-related decline in GH secretion (somatopause) is associated with increased adiposity, decreased lean mass, reduced bone density, and impaired cognitive function. GHRP-2 has been used in research to characterize the age-related changes in GHS-R1a responsiveness and to investigate whether pharmacological GH stimulation can partially reverse somatopause-associated physiological decline [22].
4. Metabolic and Obesity Research
GHRP-2's ability to stimulate GH release in obese subjects (who are typically GH-refractory due to elevated somatostatin tone and free fatty acid-mediated somatotroph suppression) makes it a valuable tool for metabolic research. Studies by Cordido and colleagues used GHRP-2 to probe the mechanisms of obesity-associated GH suppression and to investigate the metabolic consequences of restoring pulsatile GH secretion in overweight individuals [23].
5. Cardiovascular Research
The cardioprotective effects of GHS-R1a agonism represent an active area of investigation. GHRP-2 has been used in preclinical models to study the direct effects of ghrelin receptor activation on cardiac contractility, ischemia-reperfusion injury, cardiac fibrosis, and heart failure progression. These studies have revealed GHS-R1a-dependent cardioprotective mechanisms independent of GH/IGF-1 axis activation, suggesting novel therapeutic targets [20].
6. Neuroprotection and Neuroscience
Research by Frago, Granado, and colleagues demonstrated neuroprotective properties of GHRP-2 in models of cerebral ischemia, traumatic brain injury, and neuroinflammation. These studies revealed that GHRP-2 can reduce neuronal apoptosis, suppress microglial activation, and preserve blood-brain barrier integrity through mechanisms involving PI3K/Akt signaling and anti-inflammatory cytokine modulation [21].
7. GHS-R1a Receptor Pharmacology
As a high-affinity peptidyl agonist with extensive pharmacological characterization, GHRP-2 serves as a critical reference compound for GHS-R1a receptor studies. It is routinely used in radioligand binding assays (often as the unlabeled competitor against [125I]-ghrelin), functional screening assays (calcium mobilization, beta-arrestin recruitment), and structure-activity relationship campaigns aimed at developing next-generation GHS-R1a modulators [10, 11, 13].
8. Combination Peptide Research
GHRP-2 is frequently used in combination with GHRH analogs such as CJC-1295 or sermorelin to study the synergistic amplification of GH release. These combination protocols exploit the convergence of Gq/calcium and Gs/cAMP signaling pathways on the somatotroph to achieve GH responses that exceed those achievable with any single agent. Such protocols are used to study maximal pituitary capacity, to investigate the physiological ceiling of GH secretion, and to develop optimized GH stimulation paradigms for research applications [7, 18, 24].
9. Anti-Doping and Sports Science Research
Due to its potent GH-releasing properties, GHRP-2 has been added to the World Anti-Doping Agency (WADA) prohibited list as a growth hormone releasing factor. This has stimulated significant research into analytical detection methods for GHRP-2 and its metabolites in biological matrices, including mass spectrometry-based assays for urine and blood samples. These detection methods serve the broader anti-doping effort and have advanced analytical chemistry techniques for peptide detection [14].
References
[1] Bowers, C.Y., Momany, F.A., Reynolds, G.A., & Hong, A. (1984). "On the in vitro and in vivo activity of a new synthetic hexapeptide that acts on the pituitary to specifically release growth hormone." Endocrinology, 114(5), 1537-1545. DOI: 10.1210/endo-114-5-1537
[2] Bowers, C.Y. (1998). "Growth hormone-releasing peptide (GHRP)." Cellular and Molecular Life Sciences, 54(12), 1316-1329. DOI: 10.1007/s000180050257
[3] Doi, M., Sugiyama, T., Izumiyama, H., Yoshimoto, T., & Hirata, Y. (2004). "Clinical features and management of growth hormone deficiency in adults: The role of the GHRP-2 stimulation test." Internal Medicine, 43(12), 1096-1100. DOI: 10.2169/internalmedicine.43.1096
[4] Bowers, C.Y., Momany, F., Reynolds, G.A., et al. (1980). "Structure-activity relationships of a synthetic pentapeptide that specifically releases growth hormone in vitro." Endocrinology, 106(3), 663-667. DOI: 10.1210/endo-106-3-663
[5] Bowers, C.Y. (2001). "Unnatural growth hormone-releasing peptide begets natural ghrelin." Journal of Clinical Endocrinology and Metabolism, 86(4), 1464-1469. DOI: 10.1210/jcem.86.4.7399
[6] Momany, F.A., Bowers, C.Y., Reynolds, G.A., et al. (1981). "Design, synthesis, and biological activity of peptides which release growth hormone in vitro." Endocrinology, 108(1), 31-39. DOI: 10.1210/endo-108-1-31
[7] Arvat, E., Maccario, M., Di Vito, L., et al. (2001). "Endocrine activities of ghrelin, a natural growth hormone secretagogue (GHS), in humans: Comparison and interactions with hexarelin, a nonnatural peptidyl GHS, and GH-releasing hormone." Journal of Clinical Endocrinology and Metabolism, 86(3), 1169-1174. DOI: 10.1210/jcem.86.3.7314
[8] Aimaretti, G., Baffoni, C., Broglio, F., et al. (2002). "Endocrine responses to ghrelin in adult patients with isolated childhood-onset growth hormone deficiency." Clinical Endocrinology, 56(6), 765-771. DOI: 10.1046/j.1365-2265.2002.01547.x
[9] Mahajan, T. & Lightman, S.L. (2002). "A simple test for growth hormone deficiency in adults." Journal of Clinical Endocrinology and Metabolism, 87(7), 3209-3213. DOI: 10.1210/jcem.87.7.8651
[10] Bednarek, M.A., Feighner, S.D., Pong, S.S., et al. (2000). "Structure-function studies on the new growth hormone-releasing peptide, ghrelin: Minimal sequence of ghrelin necessary for activation of growth hormone secretagogue receptor 1a." Journal of Medicinal Chemistry, 43(23), 4370-4376. DOI: 10.1021/jm0001727
[11] Holst, B., Cygankiewicz, A., Jensen, T.H., Ankersen, M., & Schwartz, T.W. (2003). "High constitutive signaling of the ghrelin receptor — identification of a potent inverse agonist." Molecular Endocrinology, 17(11), 2201-2210. DOI: 10.1210/me.2003-0069
[12] Kojima, M., Hosoda, H., Date, Y., Nakazato, M., Matsuo, H., & Kangawa, K. (1999). "Ghrelin is a growth-hormone-releasing acylated peptide from stomach." Nature, 402(6762), 656-660. DOI: 10.1038/45230
[13] Howard, A.D., Feighner, S.D., Cully, D.F., et al. (1996). "A receptor in pituitary and hypothalamus that functions in growth hormone release." Science, 273(5277), 974-977. DOI: 10.1126/science.273.5277.974
[14] Ghigo, E., Arvat, E., Muccioli, G., & Camanni, F. (1997). "Growth hormone-releasing peptides." European Journal of Endocrinology, 136(5), 445-460. DOI: 10.1530/eje.0.1360445
[15] Dickson, S.L., Leng, G., & Robinson, I.C.A.F. (1993). "Systemic administration of growth hormone-releasing peptide activates hypothalamic arcuate neurons." Neuroscience, 53(2), 303-306. DOI: 10.1016/0306-4522(93)90197-N
[16] Tannenbaum, G.S. & Bowers, C.Y. (2001). "Interactions of growth hormone secretagogues and growth hormone-releasing hormone/somatostatin." Endocrine, 14(1), 21-27. DOI: 10.1385/ENDO:14:1:021
[17] Tschop, M., Smiley, D.L., & Heiman, M.L. (2000). "Ghrelin induces adiposity in rodents." Nature, 407(6806), 908-913. DOI: 10.1038/35038090
[18] Popovic, V., Damjanovic, S., Micic, D., et al. (1995). "Blocked growth hormone-releasing peptide (GHRP-6)-induced GH secretion and absence of the synergic action of GHRP-6 plus GH-releasing hormone in patients with hypothalamopituitary disconnection: Evidence that GHRP-6 main action is exerted at the hypothalamic level." Journal of Clinical Endocrinology and Metabolism, 80(3), 942-947. DOI: 10.1210/jcem.80.3.7883853
[19] Cheng, K., Chan, W.W., Barreto, A., Convey, E.M., & Smith, R.G. (1989). "The synergistic effects of His-D-Trp-Ala-Trp-D-Phe-Lys-NH2 on growth hormone (GH)-releasing factor-stimulated GH release and intracellular adenosine 3',5'-monophosphate accumulation in rat primary pituitary cell culture." Endocrinology, 124(6), 2791-2798. DOI: 10.1210/endo-124-6-2791
[20] Nagaya, N., Moriya, J., Yasumura, Y., et al. (2004). "Effects of ghrelin administration on left ventricular function, exercise capacity, and muscle wasting in patients with chronic heart failure." Circulation, 110(24), 3674-3679. DOI: 10.1161/01.CIR.0000149746.62908.BB
[21] Frago, L.M., Paneda, C., Dickson, S.L., Hewson, A.K., Argente, J., & Chowen, J.A. (2002). "Growth hormone (GH) and GH-releasing peptide-6 increase brain insulin-like growth factor-I expression and activate intracellular signaling pathways involved in neuroprotection." Endocrinology, 143(10), 4113-4122. DOI: 10.1210/en.2002-220167
[22] Bowers, C.Y., Granda, R., Mohan, S., Kuipers, J., Baylink, D., & Veldhuis, J.D. (2004). "Sustained elevation of pulsatile growth hormone (GH) secretion and insulin-like growth factor I (IGF-I), IGF-binding protein 3 (IGFBP-3), and IGFBP-5 concentrations during 30-day continuous subcutaneous infusion of GH-releasing peptide-2 in older men and women." Journal of Clinical Endocrinology and Metabolism, 89(5), 2290-2300. DOI: 10.1210/jc.2003-031799
[23] Cordido, F., Penalva, A., Dieguez, C., & Casanueva, F.F. (1993). "Massive growth hormone (GH) discharge in obese subjects after the combined administration of GH-releasing hormone and GHRP-6: Evidence for a marked somatotroph secretory capability in obesity." Journal of Clinical Endocrinology and Metabolism, 76(4), 819-823. DOI: 10.1210/jcem.76.4.8473388
[24] Popovic, V., Leal, A., Micic, D., et al. (2000). "GH-releasing hormone and GH-releasing peptide-6 for diagnostic testing in GH-deficient adults." Lancet, 356(9236), 1137-1142. DOI: 10.1016/S0140-6736(00)02755-0
[25] Pihoker, C., Kearns, G.L., French, D., & Bowers, C.Y. (1998). "Pharmacokinetics and pharmacodynamics of growth hormone-releasing peptide-2: A phase I study in children." Journal of Clinical Endocrinology and Metabolism, 83(4), 1168-1172. DOI: 10.1210/jcem.83.4.4731
Disclaimer
This article is for educational and informational purposes only. It is not intended as medical advice, diagnosis, or treatment recommendation. GHRP-2 (pralmorelin) is sold as a research peptide and is not approved for human therapeutic use outside of its diagnostic indication in Japan. The information presented herein is derived from published peer-reviewed scientific literature and does not constitute a recommendation for any specific research protocol or application. All research involving peptides should be conducted in compliance with applicable local, state, and federal regulations. Researchers should consult relevant institutional review boards and regulatory bodies before initiating any research protocols.
Published by BLL Peptides — Premium Research Peptides
GHRP-2 is a research-grade synthetic growth hormone secretagogue studied for potent GH-releasing effects with minimal desensitization, making it valuable for HPA axis and somatotropic axis research. Researchers investigating growth hormone signaling and anabolic pathways rely on pharmaceutical-grade purity for reproducible results. Available at BLL Peptides — USA-made, rigorously tested.
| ✅ COA tested every batch | ✅ 98%+ purity guaranteed |
| ✅ USA manufactured, GMP-certified | ✅ Glass vials — not plastic |
| ✅ Veteran-owned company | ✅ Free shipping over $150 |


Related Products
-
Oxytocin 2mg (3ml)
Original price was: $149.00.$59.99Current price is: $59.99. -
Epithalon 10mg (3ml)
Original price was: $99.99.$49.99Current price is: $49.99. -
DSIP 10mg (3ml)
Original price was: $120.00.$69.99Current price is: $69.99. -
BPC-157 10mg (3ml)
Original price was: $95.00.$54.99Current price is: $54.99.